AmplifX : Aide rapide
mardi 26 octobre 2004.A quoi sert AmplifX
AmplifX est initialement destiné à rechercher dans une collection d’amorces, telle que tout biologiste moléculaire en possède dans ces frigos, ceux qui peuvent servir à amplifier un fragment d’une séquence cible, par exemple, et tout particulièrement, pour dessiner des stratégies de cribles de transformants par PCR. Un certain nombre d’informations sont associés à ces amorces : certaines calculées par AmplifX comme leur température de fusion (TM) ou leur qualité et d’autres ajoutées par l’utilisateur (commentaires) De nouvelles amorces peuvent aussi être calculées par AmplifX en réglant plusieurs paramètres.Remarque sur les possibilités d’AmplifX
Ce programme n’a pas une ambition de programme commercial. En cela certains algorithmes ne sont pas les plus performants du moment et certaines aspects de l’interface ne sont pas dans une finition impeccable (n’hésitez pas toutefois à en faire la remarque ; tout peut être amélioré : c’est une question d’investissement !)Informations légales Ce programme est mis gratuitement à la disposition de la communauté scientifique en l’état et tel quel. L’auteur ne pourra être tenu responsable d’un éventuel mauvais fonctionnement. Les utilisateurs sont très fortement invités à « s’enregistrer » en remplissant le formulaire en ligne (accessible par le menu Aide) afin de pouvoir être rapidement informé de toute mise à jour importante.
Comment utiliser AmplifX
- La Séquence cible La fenêtre d’AmplifX est organisée en deux parties : la partie supérieure contient la zone textuelle, elle même organisée en trois panneaux : “Séquence” , “Liste d’amorces” et “Infos”.
Le panneau “Séquence” est la zone dans laquelle est affichée la séquence qui servira de matrice pour la PCR. On peut ouvrir une séquence aux formats DNA Strider’, GeneBank’, EMBL’, GCG’ ou Fasta’ ainsi que n’importe quel format texte qui sera alors reconnu comme format Brut’ et pour lequel seuls les caractères autorisés seront pris en compte. On peut aussi coller directement, ce qui est souvent la solution la plus pratique lorsque on utilise une application spécifique pour manipuler les séquences (telles que Gene Construction Kit, MacVector,...), AmplifX élimine les caractères différents de “ A, T/U, C, G”. On peut donc directement copier (ou ouvrir un fichier texte) à partir d’une source contenant, par exemple, des espaces, des nombres, etc. Par contre AmplifX ne tient pas compte des nucléotides dégénérés (IUPAC : N, S, W, ...).
Il peut être utile de spécifier certaines zones d’intérêt tout simplement en alternant minuscules et majuscules (voir le paragraphe concernant la représentation graphique de la séquence cible)
- La liste d’amorces Le panneau “Liste d’amorces” contient la liste de primers et est associé au menu “Amorces”. Pour importer une liste d’amorces, il faut utiliser le menu du même nom. Il permet d’importer un fichier au format texte. Celui-ci doit être formaté comme suit : une ligne par primer avec : la séquence du primer, son nom ou ID, son nom descriptif ou catégorie (Il peut y avoir d’autres éléments qui suivent mais ceux-ci ne seront pas pris en compte). Chaque item est séparé par une tabulation. Ce type de fichier est facilement créé à partir d’un tableur ou d’un programme de base de données, par exemple. On peut aussi saisir directement les primers dans AmplifX en créant une ligne vide par le menu « Ajouter un primer ».
AmplifX calcule un score de qualité basé sur un certain nombre de caractéristiques telles que le TM, la composition GC, la stabilité 3’, la complexité (polyX et répétition de triplet) et les formations de dimères.
Le calcul de « TM » est effectué avec trois méthodes différentes : une méthode simple et très approximative : TM=81.5+0.41*GC-675/N, une autre tenant compte de la concentration en sel dans une PCR TM = 81.5 + 16.6 x log10([Na+]+[K+])+0.41 x(%GC) - 675/N (avec par défaut dans le programme : [Na+]+[K+]=0.05 (50mM)) enfin la dernière qui est la plus précise à l’heure actuelle dite « base stacking method » :TM= ??H/( ??S+0.368xNxln[Na+]+ R x ln[Primer]/4) avec R=1.987 et les différents ?H et ?S [1]
La liste peut être triée par un clic dans les en-têtes. Il est conseillé d’utiliser dans la colonne« ID » des données d’une forme triable de manière alphanumérique. Par exemple si vous souhaitez y mettre un ID de type numéro de série, pensez à formater ces numéros avec un nombre constant de chiffre : Pr0001, Pr0002,... (Les colonnes Qualité et longueur, calculées par le programme sont triables sur la valeur du nombre)
La colonne « Nom descriptif » peut aussi être employé pour créer des sous-sections en plaçant au début des données, par exemple on peut vouloir faire une catégorie « SYNT » pour les amorces contenant des parties synthétiques. cela permettra ensuite de rapidement les retrouver par simple classement sur cette colonne. Lorsque l’on double-clique sur une ligne, soit on édite la cellule (Séquence, ID, nom descriptif) soit on sélectionne la ligne entière ce qui entraîne l’apparition d’une fenêtre tiroir contenant des détails sur la qualité de l’amorce et un champ commentaire qui peut être utilisé pour tout usage (nom du possesseur de l’amorce, congélateur et boite de rangement, commentaire sur son utilisation,...)
La liste peut être sauvée dans un format spécifique (AmplifX primer list) qui est du texte brut structuré de façon spécifique (ne le modifier pas avec un éditeur de texte).
Un bouton en haut à gauche permet de définir un fichier précédemment sauvé comme fichier par défaut , qui se charge automatiquement à l’ouverture du programme.
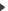 Le panneau informations
Le panneau infos donne les infos pour chaque élément graphique cliqué. (Voir ci-dessous). Les données peuvent y être copié de façon classique.
Le panneau informations
Le panneau infos donne les infos pour chaque élément graphique cliqué. (Voir ci-dessous). Les données peuvent y être copié de façon classique.
A propos de la représentation graphique
La partie graphique contient la séquence cible représentée par une ligne, colorée en deux teintes (une pour les nucléotides saisis en minuscules et une autre pour ceux saisis en majuscules) au dessus et au dessous apparaissent les amorces qui hybrident sur la séquence. Celles ci sont représentés par des symboles orientés pleins ou vides en fonction de l’efficacité supposé de l’amorçage. (Le seuil des scores pour chaque catégorie sont modifiables dans la fenêtre Préférences) Le survol sur ces symboles fait apparaître le nom de l’amorce et un clic dessus donne, dans le panneau “infos”, les différentes informations concernant l’amorce et les caractéristiques de l’hybridation. (attention les TM indiqués sont ceux du primer sans tenir compte de la qualité de l’hybridation, en effet il n’existe pas encore de méthode valable pour les appariements imparfaits au delà d’une base de non appariement.)Au dessous des lignes figurent les séquences amplifiées. Leur survol indique la taille du fragment et le clic renseigne le panneau “infos”. Le clic permet aussi de sélectionner directement la portion de séquence amplifiée (La vraie séquence amplifiée, c’est à dire celle qui contient les éventuelles modifications apportées par les amorces est automatiquement copiée dans le presse-papier)
Pour changer l’échelle de la représentation graphique il faut utiliser les petits boutons « + » , « -« et « 0 » qui permettent de zoomer, de « dézoomer » ( !) ou encore de revenir à la vue initiale. Une manière plus pratique pour restreindre la vue à une zone d’intérêt et de « drager » la dite zone tout en tenant la touche Majuscule. L’ascenseur horizontal permet de déplacer la portion visible.
Il est possible d’imprimer ou d’exporter la partie visible par les menus appropriés du menu « Fichier ».
Quelques menus particuliers
Le menu préférences
Il permet de spécifier un fichier de primer par défaut au lancement de AmplifX. Ce fichier doit être une liste de primer qui a été précédemment importé et sauvé au format “AmplifX primer list”. Certaines constantes utilisées pour les détections d’hybridation des primers sur la séquence cible peuvent être modifiées. Ces constantes sont commentées dans la fenêtre Préférences.
Le menu “Chercher”
dans le menu “Edition” permet de rapidement localiser et sélectionner des amorces dont les différents champs (nom ou description) contiennent une chaîne de caractères donnée. Les primers ainsi sélectionnées peuvent être ainsi rapidement cochés pour être utilisé dans la « PCR virtuelle » manuellement ou automatiquement par le menu « Cocher les primers trouvés ».
La recherche peut aussi s’effectuer sur la séquence cible (en cochant le bouton radio approprié) afin de par exemple localisé un motif spécifique. (Il est alors possible de le mettre en évidence par le jeu des minuscules/majuscule, voir le menu « Edition »)
Le menu « coller le brin complémentaire »
est utile pour le design « à la volée » par sélection directe dans le champ « séquence », le calcul de la qualité y est effectuée en direct. On peut alors copier la sélection courante et la coller soit directement soit par ce menu dans une nouvelle ligne de la liste d’amorces.
Dessiner des amorces avec AmplifX
Bien que sa première finalité soit de tester rapidement des amorces déjà existantes, AmplifX incorpore depuis la version 1.1 une fonction de recherche et de design d’oligos.Cette fonction est accessible par le menu « Amorces « -> »Dessiner des amorces ». Une nouvelle fenêtre s’ouvre alors qui permet de régler certains paramètres spécifiques à cette fonction, tels que la longueur des amorces souhaitée, la différence de TM entre les deux et le score de qualité minimum servant de seuil. Ce calcul de qualité est lui même dépendant de certains paramètres qui sont modifiables par le menu « Préférences », tels que par exemple, le TM ou la composition GC. On doit ensuite renseigner de façon plus ou moins exhaustive en fonction de ce que l’on souhaite les zones de recherche des amorces sens et anti-sens et la longueur du produit d’amplification (si une partie des informations n’est pas fournis le programme la calcule automatiquement avec plus ou moins de succès) On peut aussi choisir de ne chercher qu’une seule amorce qui fonctionne avec une déjà définie. On peut alors, par exemple, coller à partir de la liste d’amorces de la fenêtre principale dans le champ « utiliser cette amorce » sens ou anti-sens. Une fois la recherche terminée on peut envoyer les amorces sélectionnées par un double-clic sur la liste proposée suivi du clic sur le bouton « Ajouter à la liste d’amorces »
Un conseil : ne spécifier pas une zone de recherche trop grande pour chaque amorce (restreindre à une zone de 100-200 bp est raisonnable), et commencer en réglant le seuil de qualité à une valeur élevé : 90. Si vous ne trouvez rien diminuer ensuite les contraintes.
Si vous voulez calculer des amorces de façon plus sophistiquée, je vous conseille une application spécialisée telle que Primer3 [2] , (une bonne implémentation de ce programme peut être trouvée à l’adresse suivante : http://frodo.wi.mit.edu/)
N’hésitez pas à m’aider à améliorer AmplifX et cette aide en m’envoyant vos commentaires à jullien.n at jean-roche dot univ-mrs dot fr
[1] Santalucia J. (1998) PNAS 95 pp1460-1465
[2] Steve Rozen and Helen J. Skaletsky (2000) Primer3 on the WWW for general users and for biologist programmers. In : Krawetz S, Misener S (eds) Bioinformatics Methods and Protocols : Methods in Molecular Biology. Humana Press, Totowa, NJ, pp 365-386