AmplifX quick help
Thursday 28th October 2004.AmplifX main purposes
The main purpose of AmplifX is to seek in a collection of primers, such as any molecular biologist get in his refrigerators, those which can be use to amplify a fragment into a target sequence, for example, and particularly, to design strategies to screen recombinant clones by PCR.Some informations are associated with each primer, some automatically computed by AmplifX (like TM, Quality, length) and other given by the user (name, comments,...) This allows general aspects of primer management (sequences and real tubes).
AmplifX can also design new primers.
Remark on the possibilities of AmplifX
This program does not have an ambition of commercial program. From this point of view it contains some algorithms that are not the most powerful of the moment and certain aspects of the interface are not in an perfect completion (do not hesitate however to make the remark of it; all can be improved: it is a question of investment!)Legal informations
This program is provided free for the scientific community ”as this”. The author couldn’t be responsible for any bad working. Users are strongly invited to register themselves with the online form accessible by the help menu. This will assure you to be kept informed for new updates and important informations about this program as soon as they are available.How using AmplifX
- The target sequence The main window of AmplifX is organized in two parts: the upper part contains the textual zone, it is organized itself in three panels: "Target", "Primer List" and "Infos". The panel "target" is the zone in which the target sequence is loaded. one can paste the sequence on which one wants to seek the matching primers. When pasting, AmplifX eliminate all the characters different from : "A, T/U, C, G,". One can thus directly copy from a source containing, for example, spaces, numbers, ... On the other hand AmplifX does not hold account of degenerate nucleotides (IUPAC: N, S, W...). AmplifX can open some file formats that are in this current version : DNA Strider’, GeneBank’, EMBL’, GCG’, Fasta’ and all pure text format that are processed like the paste command (paste only authorized characters) It can be useful to mark some regions of special interest by alternating upper and lower cases characters into the sequence (see the paragraph relating to the graphical representation of the target sequence)
-The Primer list The panel "Primer List" contains the list of primers and is associated to the " Primers" menu. To import a primer list, use the menu of the same name. It makes it possible to import a file in text format. This one must be formatted as follows: one line by primer with: the primer sequence, its name or ID, its descriptive name or category (There can be others elements which follow but those will not be taken into account). Each item is separated from the others by a tabulation. This type of file is easily created with Excel or FileMaker, for example. One can also directly enter the primers in AmplifX by creating a blank line with the menu “Add a new primer”.
AmplifX calculates a Quality score, computing some characteristics like TM, GC composition, complexity (polyX and triplet repetitions), 3’ stability and self dimers. (the best is 100, the worst is 0)
The “TM” value is computed with three different methods: a simple and very and approximate method: TM=81.5+0.41*GC-675/N, another holding account of the salt concentration in a PCR TM = 81.5 +16.6 X log10([Na+]+[K+])+0.41 x(%GC) - 675/N (with default values: [ Na+]+[K+]=0.05 (50mM)) finally the last one which is the most precise a that time called :"bases stacking method” : TM=dH/(dS+0.368xNxln[Na+]+ R x ln[Primer]/4) with R=1.987 and the different dH and dS taken in [1]
The list can be sorted by clicking in the headings. It is advised to use data sortable in an alphanumeric way in the column “ID”. For example if you wish to use serial numbers, think of formatting these numbers with a constant number of digits : Pr0001, Pr0002... (the columns Quality and length,calculated by the program are sortable by numerical value) the "descriptive name" column can also be employed to create sub-sections while placing special strings at the beginning of the field, for example one can want to make a category "SYNT" for the primers containing synthetic parts. this will make it possible to quickly find them by simple sorting on this column.
When one double-clicks on a line, either one edit the cell (Sequence, ID, descriptive name) or one selects the whole line. This involves the appearance of a window drawer containing some details about the quality of the primer and a field “comments” which can be used for any use (name of the owner of the primer, freezer and box, comment on its use...)
The primer list can be saved in a specific format (AmplifX primer list) which is a text file structured in a specific way (it can be opened by any text editor but don’t modify it manually). A button in top on the left makes it possible to define a file previously saved as a default file, i.e. automaticaly loaded at AmplifX start up.
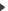 The informations panel :
The informations panel :
The panel infos is self explanory and gives the informations for each clicked graphical element (See below). Its content can be recovered by classic “copy” command.
The PCR chart
The graphic part contains the target sequence drawn as a simple line in two colors relative to positions of upper and lower cases nucleotide characters. At the top and below of this line hybridizing primers appear as two oriented symbols according to the direction of the hybridized strand. These symbols are drawn filled or empty property regarding to the strength of the match. (The match cut-off scores can be changed by the preference menu).
The mouse standing onto these symbols let appear the name of the primers and a click above gives, in the panel "infos", the different informations concerning the primer and some hybridization characteristics (Be careful : given TM are those of the primer without holding account of the hybridization quality indeed it does not exist yet method valid for imperfect pairings.)
In the lower part appear the amplified sequences. Their overflight indicates the size of the fragment and the click informs the panel "infos". The click allows also to select directly the portion of the amplified sequence and automaticaly paste the real amplified fragment (i.e. with the primer sequences at the boundaries) into the clipboard (useful to copy-paste in another application).
To change the graphic drawing scale one can use the small buttons "+ ", “-“" and “0” which allow plus zoom, minus zoom or to restore the initial scale. A more practical way to restrict the view to a region of interest is to drag the mouse while pressing the Shift key. An horizontal scroll bar allow the moving of the viewable portion.
The currently viewable portion of the chart can be printed or exported as standard graphic format by the way of the two dedicated items in the “file” menu
About some menus of special interest :
The Preferences Menu
It allows the specification of a Primer List file to be open by default.
This one have to be already save with AmplifX. Some constants used for the detection calculations can be modified to allow more sensitive or faster searches. (they are commented into the Pref window)
The search menu :
A function "Search" in the "Edition" menu makes it possible to quickly locate and select primers whose name or comment fields contain a given string. The found primers can thus quickly checked for use into the virtual PCR manually or automatically by the menu "Check found primers”. The search can be done into the target sequence also by clicking the appropriate radio buttion.
The "Paste complementary strand" menu
It is useful for the primer design by direct selection in the "Target"field. In which the calculation of quality is carried out “live” as selection change. One can then copy the current selection and paste it either directly or by this menu in a new line of the primer list.
Primer design with AmplifX
Although its first finality is to test already existing primers quickly, AmplifX incorporates since version 1.1 a function of primer design. This function is accessible by the menu "Primers" - > "Design primers". A new window opens which makes it possible to fix some specific parameters to this function, such as the length of the primers wished, the difference of TM between the two ones and the minimum score of quality being used as threshold. This calculation of quality is also dependent on certain parameters which can be changed by the menu " Preferences", such as for example, TM or GC composition.
One must then fill in a more or less exhaustive way according to what one wishes : the forward and reverse primer searching zones and the length of the product of amplification (if a part of information is not provided the program automatically calculates it with more or less success) One can choose to search for only one primer by filling one of the two “Use this primer” fields.
Once the search is finished, one can send the primers selected by double-clicking onto the list and clicking on the button "Add to primer list"
Tip : Do not specify a large searching zone for each primer (restricting to 100-200 bp is reasonable), and start by fixing the quality score threshold to a high value ( arround 90). If no primer pairs were found, decrease the constraints then.
If you want to calculate primers in more sophisticated way, I advise you to use a specialized application such as Primer3 [2]), (a good implementation of this program can be found at the following address:http://frodo.wi.mit.edu/)
Do not hesitate to help me to improve AmplifX and this help by sending your comments to jullien.n at jean-roche dot univ-mrs dot fr
[1] Santalucia J PNAS 95 pp1460-1465 (1998)
[2] Steve Rozen and Helen J. Skaletsky (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press, Totowa, NJ, pp 365-386